Estimate high-dimensional additive models using spectral deconfounding scheidegger2023spectralSDModels. The covariates are expanded into B-spline basis functions. A spectral transformation is used to remove bias arising from hidden confounding and a group lasso objective is minimized to enforce component-wise sparsity. Optimal number of basis functions per component and sparsity penalty are chosen by cross validation.
SDAM(
formula = NULL,
data = NULL,
x = NULL,
y = NULL,
Q_type = "trim",
trim_quantile = 0.5,
q_hat = 0,
nfolds = 5,
cv_method = "1se",
n_K = 4,
n_lambda1 = 10,
n_lambda2 = 20,
Q_scale = TRUE,
ind_lin = NULL,
mc.cores = 1,
verbose = TRUE
)Arguments
- formula
Object of class
formulaor describing the model to fit of the formy ~ x1 + x2 + ...whereyis a numeric response andx1, x2, ...are vectors of covariates. Interactions are not supported.- data
Training data of class
data.framecontaining the variables in the model.- x
Matrix of covariates, alternative to
formulaanddata.- y
Vector of responses, alternative to
formulaanddata.- Q_type
Type of deconfounding, one of 'trim', 'pca', 'no_deconfounding'. 'trim' corresponds to the Trim transform Cevid2020SpectralModelsSDModels as implemented in the Doubly debiased lasso Guo2022DoublyConfoundingSDModels, 'pca' to the PCA transformationPaul2008PreconditioningProblemsSDModels. See
get_Q.- trim_quantile
Quantile for Trim transform, only needed for trim, see
get_Q.- q_hat
Assumed confounding dimension, only needed for pca, see
get_Q.- nfolds
The number of folds for cross-validation. Default is 5.
- cv_method
The method for selecting the regularization parameter during cross-validation. One of "min" (minimum cv-loss) and "1se" (one-standard-error rule) Default is "1se".
- n_K
The number of candidate values for the number of basis functions for B-splines. Default is 4.
- n_lambda1
The number of candidate values for the regularization parameter in the initial cross-validation step. Default is 10.
- n_lambda2
The number of candidate values for the regularization parameter in the second stage of cross-validation (once the optimal number of basis function K is decided, a second stage of cross-validation for the regularization parameter is performed on a finer grid). Default is 20.
- Q_scale
Should data be scaled to estimate the spectral transformation? Default is
TRUEto not reduce the signal of high variance covariates.- ind_lin
A vector of indices specifying which covariates to model linearly (i.e. not expanded into basis function). Default is `NULL`.
- mc.cores
Number of cores to use for parallel processing, if
mc.cores > 1the cross validation is parallelized. Default is `1`. (only supported for unix)- verbose
If
TRUEfitting information is shown.
Value
An object of class `SDAM` containing the following elements:
- X
The original design matrix.
- p
The number of covariates in `X`.
- intercept
The intercept term of the fitted model.
- K
A vector of the number of basis functions for each covariate, where 1 corresponds to a linear term. The entries of the vector will mostly by the same, but some entries might be lower if the corresponding component of X contains only few unique values.
- breaks
A list of breakpoints used for the B-splines. Used to reconstruct the B-spline basis functions.
- coefs
A list of coefficients for the B-spline basis functions for each component.
- active
A vector of active covariates that contribute to the model.
References
See also
Examples
set.seed(1)
X <- matrix(rnorm(10 * 5), ncol = 5)
Y <- sin(X[, 1]) - X[, 2] + rnorm(10)
model <- SDAM(x = X, y = Y, Q_type = "trim", trim_quantile = 0.5, nfold = 2, n_K = 1)
#> [1] "Initial cross-validation"
#> [1] "Second stage cross-validation"
# \donttest{
library(HDclassif)
#> Loading required package: MASS
data(wine)
names(wine) <- c("class", "alcohol", "malicAcid", "ash", "alcalinityAsh", "magnesium",
"totPhenols", "flavanoids", "nonFlavPhenols", "proanthocyanins",
"colIntens", "hue", "OD", "proline")
wine <- log(wine)
# estimate model
# do not use class in the model and restrict proline to be linear
model <- SDAM(alcohol ~ -class + ., wine, ind_lin = "proline", nfold = 3)
#> [1] "Initial cross-validation"
#> [1] "Second stage cross-validation"
# extract variable importance
varImp(model)
#> class malicAcid ash alcalinityAsh magnesium
#> 2.200743e-04 6.616034e-06 1.077807e-05 0.000000e+00 0.000000e+00
#> totPhenols flavanoids nonFlavPhenols proanthocyanins colIntens
#> 0.000000e+00 3.574768e-06 5.505266e-06 2.152082e-07 4.544776e-04
#> hue OD proline
#> 0.000000e+00 0.000000e+00 0.000000e+00
# most important variable
mostImp <- names(which.max(varImp(model)))
mostImp
#> [1] "colIntens"
# predict for individual Xj
predJ <- predict_individual_fj(object = model, j = mostImp)
plot(wine[, mostImp], predJ,
xlab = paste0("log ", mostImp), ylab = "log alcohol")
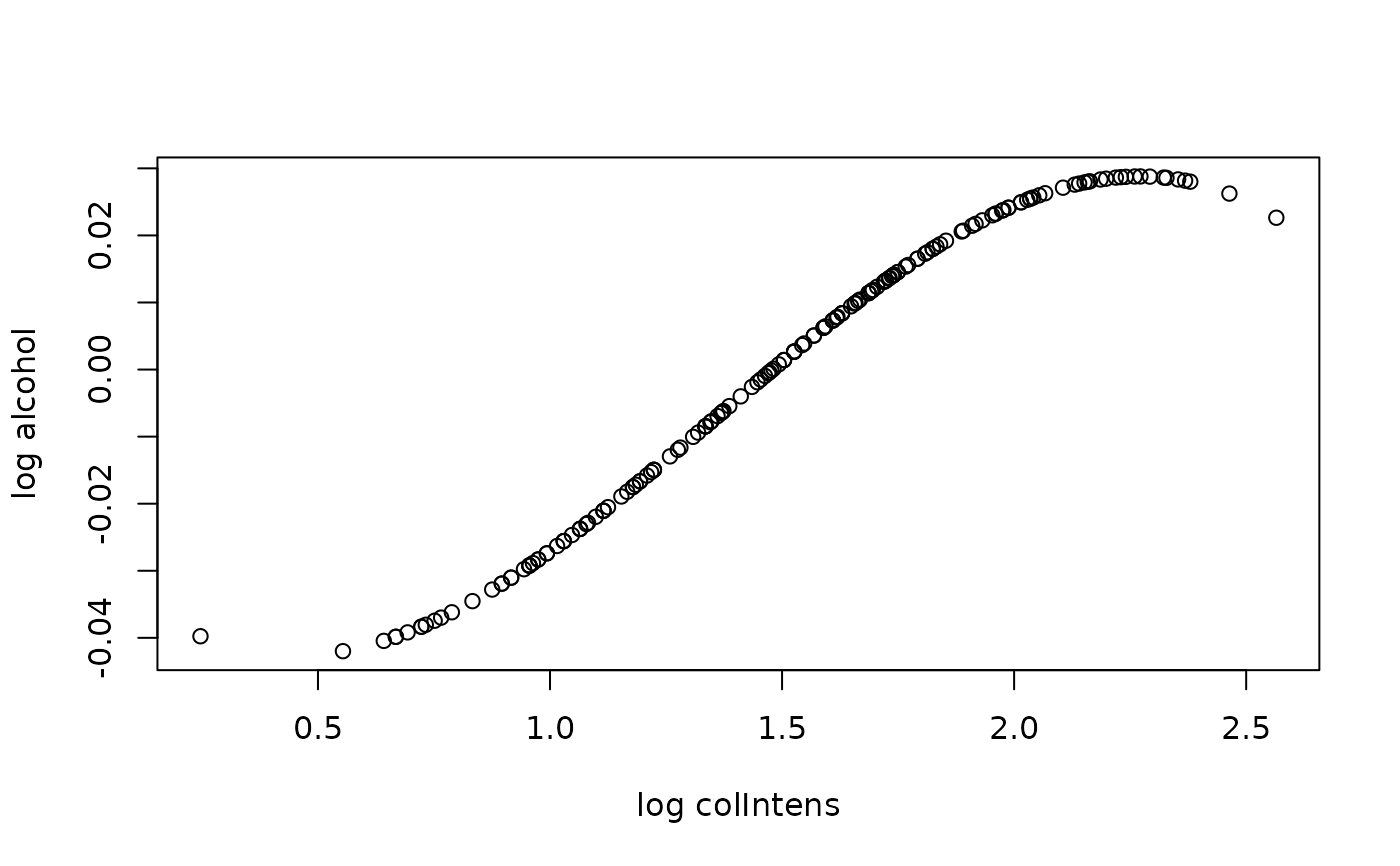 # partial dependece
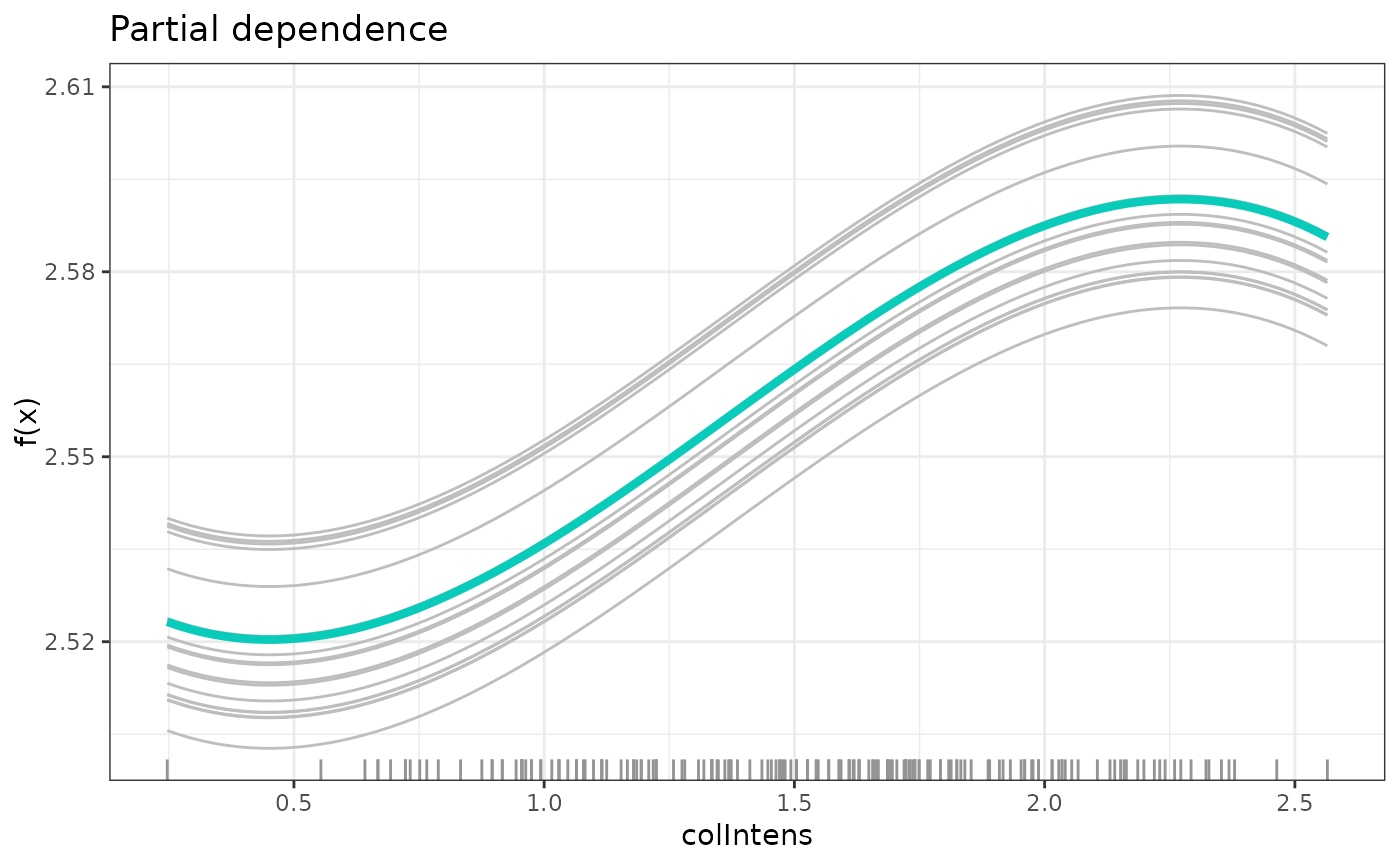
plot(partDependence(model, mostImp))
# partial dependece
plot(partDependence(model, mostImp))
 # predict
predict(model, newdata = wine[42, ])
#> [1] 2.574795
## alternative function call
mod_none <- SDAM(x = as.matrix(wine[1:10, -c(1, 2)]), y = wine$alcohol[1:10],
Q_type = "no_deconfounding", nfolds = 2, n_K = 4,
n_lambda1 = 4, n_lambda2 = 8)
#> [1] "Initial cross-validation"
#> [1] "Second stage cross-validation"
# }
# predict
predict(model, newdata = wine[42, ])
#> [1] 2.574795
## alternative function call
mod_none <- SDAM(x = as.matrix(wine[1:10, -c(1, 2)]), y = wine$alcohol[1:10],
Q_type = "no_deconfounding", nfolds = 2, n_K = 4,
n_lambda1 = 4, n_lambda2 = 8)
#> [1] "Initial cross-validation"
#> [1] "Second stage cross-validation"
# }